
GDEC Sample Submission Guide:
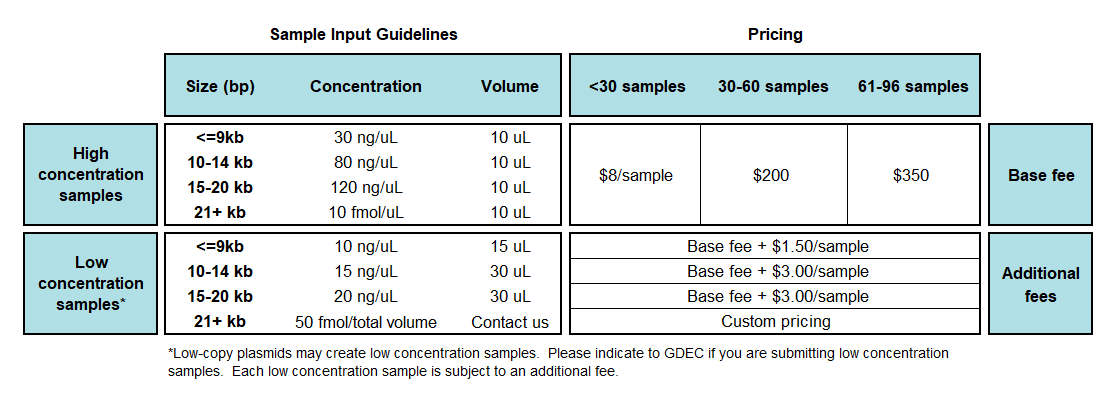
Instructions:
-
Register with GDEC and FOM.
Find instructions here. -
Set up sequencing reactions according to the guidelines above.
If you have <=24 samples, submit in 8-well PCR strip-tubes, numbered 1 through 24. For >24 samples, submit in a PCR plate, setup by COLUMN such that samples 1, 2, 3 correspond to positions A1, B1, C1. -
Fill in the WPS Sample Submission Form.
Email the completed form to gdec@rice.edu. -
Label your sample submission: Date (YYMMDD) - User Name.
The date is the drop-off date at GDEC. The user name is the name of the person whose account we are using. The data will be sent to that person. -
Drop off samples at the GDEC lab (BRC 215).
We have a sample drop box outside the lab. Please let us know if any of your samples are nonstandard (low concentration, etc.). This helps us prepare the library accordingly.
Turnaround is 2-3 days, generally.
Data will be sent to the user via a Google Drive link. The data will be deleted after 3 weeks, so please download your files for long term storage.
GDEC Users: Please remember to acknowledge the use of GDEC instrumentation, services, and support in your presentations and publications. Include the following statement:
“This work was supported by the Genetic Design and Engineering Center (GDEC) at Rice University, which is funded by CPRIT RP210116.”
F.A.Q.'s:
-
What data is provided by GDEC?
-
Your whole plasmid sequencing data is shared in a Google Drive folder named according to the drop-off date. The folder has several files:
fastq_pass: These are all of your raw fastq files sorted by barcode. This includes an initial quality filtering step that removes low quality “failed” reads.
consensus_fasta: This is the consensus sequence produced by our bioinformatic pipeline.
consensus_genbank: This is the same sequence as the consensus_fasta but includes annotations from the pLannotate program.
consensus_fastq: This is the same sequence as the consensus_fasta but includes quality information about each base.
read_length_histogram: This is a histogram plotting the lengths of each read (trimmed fastq_pass reads). The estimated plasmid size is plotted as a vertical line.
wf-clone-validation-report: This is a report generated by ONT’s Epi2me wf-clone-validation software that we use to create consensus sequences. This file includes a preview of the sample, a read length histogram, quality score histogram, and reasons for assembly failure if no consensus is formed. At GDEC we run the wf-clone-validation program two times to try to rescue failed assemblies, so there are sometimes two files included here.
consensus_quality_check: This is a file that compares the predicted plasmid size (inferred from the read_length_histogram) to the length of the consensus sequence. In some instances the consensus sequence does not match the known size of the plasmid, so this file helps to quickly flag such samples.
- Why does the consensus sequence not match the predicted plasmid size?
-
Usually this is caused by the consensus sequence containing an erroneous deletion. This is particularly likely to happen if the plasmid contains repeating sequences or does not have enough long reads. If you suspect that the consensus is wrong, we suggest you take your raw fastqs and align them to a reference. Other error modes are that the consensus can be too long, or that the predicted plasmid size was incorrectly estimated. You can manually inspect the read length histogram to see if the estimated plasmid size is incorrect.
- How can I investigate the quality of each base using the .fastq consensus?
-
Nanopore accuracy may vary on a per base basis. For example, accuracy may drop in homopolymer regions (i.e. regions with the same base repeated 5+ times) or in regions with high amounts of secondary structure. The consensus .fastq file provides information about the quality of a particular base in a sequence. The quality can be manually interpreted from the .fastq file or visualized in a viewer such as SnapGene.
- How can I do an alignment with raw .fastq files?
-
To align .fastq files to a .fasta reference you can use ONT’s free bioinformatics suite called EPI2ME available on their website.
- Why are there multiple peaks in the read length histogram?
-
The first peak is usually short reads. While some amount of short reads is always to be expected, a large fraction of short reads indicates DNA degradation. Typically there is a second, high peak which is likely created by linearized plasmid (where we calculate the expected plasmid size). Then there is normally a small peak 2X larger than that, which can indicate plasmid concatemerization. Other large peaks may indicate sample contamination with other plasmids.
Some samples have a high proportion of long reads (10k+) which do not correspond to their plasmid. In many cases this is caused by purifying plasmid from a strain that contains the EndA1 gene. These EndA1+ strains are inappropriate for preparing samples for nanopore analysis. For example, BL21 and MG1655 are NOT appropriate for WPS.
- Can I pool multiple plasmids together into one sample?
-
Yes, but our consensus algorithm will not produce an accurate result. Also, keep in mind we typically achieve 100-500 passed reads per plasmid so for deep sequencing, you need to divide your sample across multiple barcodes.
- Why did my sequencing fail to produce a consensus file?
-
We cannot guarantee that a consensus will be formed for sequencing. The main reason for failure is insufficient reads but it can also be caused by sample contamination or complexities in the sequence (numerous repeats, large size, etc.).
- Why did my sequencing produce insufficient reads?
-
If your sequencing did not produce enough reads it typically means that your sample did not have enough long, high quality DNA to begin with. We see this issue most often with low copy plasmids that seem to have a lower fraction of full length plasmid to “junk” DNA including degraded plasmid or genomic contamination. For these samples we suggest submitting higher volumes (>=15 uL) or doubling the concentration in those samples.
Other reasons for not getting enough reads include low quality sample preparation. DNA should be prepared from an appropriate cloning strain (with endA1 deleted) such as DH10B or Dh5a. DNA should be prepared from fresh cell pellets that have not undergone excessive cold storage. Excessive cell lysis during plasmid prep can cause genomic and plasmid fragmentation.
Plasmids that have been stored improperly are also prone to degradation which can lead to short reads. Ensure plasmids are stored at -20 ºC in sterile, nuclease-free water or buffer.
Other reasons for a lack of reads could be that the plasmid was larger than expected and that not enough was loaded for the size of the plasmid (see GDEC Sample Submission Guide above).